Introduction
Next-Generation Sequencing (NGS) has enabled researchers to explore genome-wide patterns with unprecedented precision. From base-level resolution to whole-genome comparisons, the demand for accuracy in sequencing outputs has never been higher. A central component in most NGS workflows is the use of polymerase chain reaction (PCR) to amplify input DNA prior to sequencing. Yet, standard PCR introduces a risk: polymerase-induced errors.
PCR errors can lead to artificial mutations, allelic imbalances, and misinterpretation of variant frequencies, which misleads data interpretation, especially in studies with rare variants or low-input DNA. This article explores how high-fidelity enzymes, designed with intrinsic proofreading capacity, can significantly mitigate these problems and ensure more trustworthy data in sequencing workflows.
Mechanisms of PCR-Induced Errors in Library Preparation
PCR-induced errors are not random; they typically arise due to:
-
Misincorporation of incorrect nucleotides.
-
Template switching under high-cycle conditions.
-
Chimeric sequence generation.
-
GC bias, where high-GC content regions are either underrepresented or distorted.
According to NCBI, these errors can become particularly problematic during the pre-amplification and final library enrichment stages, where input DNA is limited and cycle numbers are high.
Even minor inaccuracies introduced early in the process are amplified exponentially, resulting in an error-rich final library. These accumulated errors are indistinguishable from real variants unless special strategies are adopted.
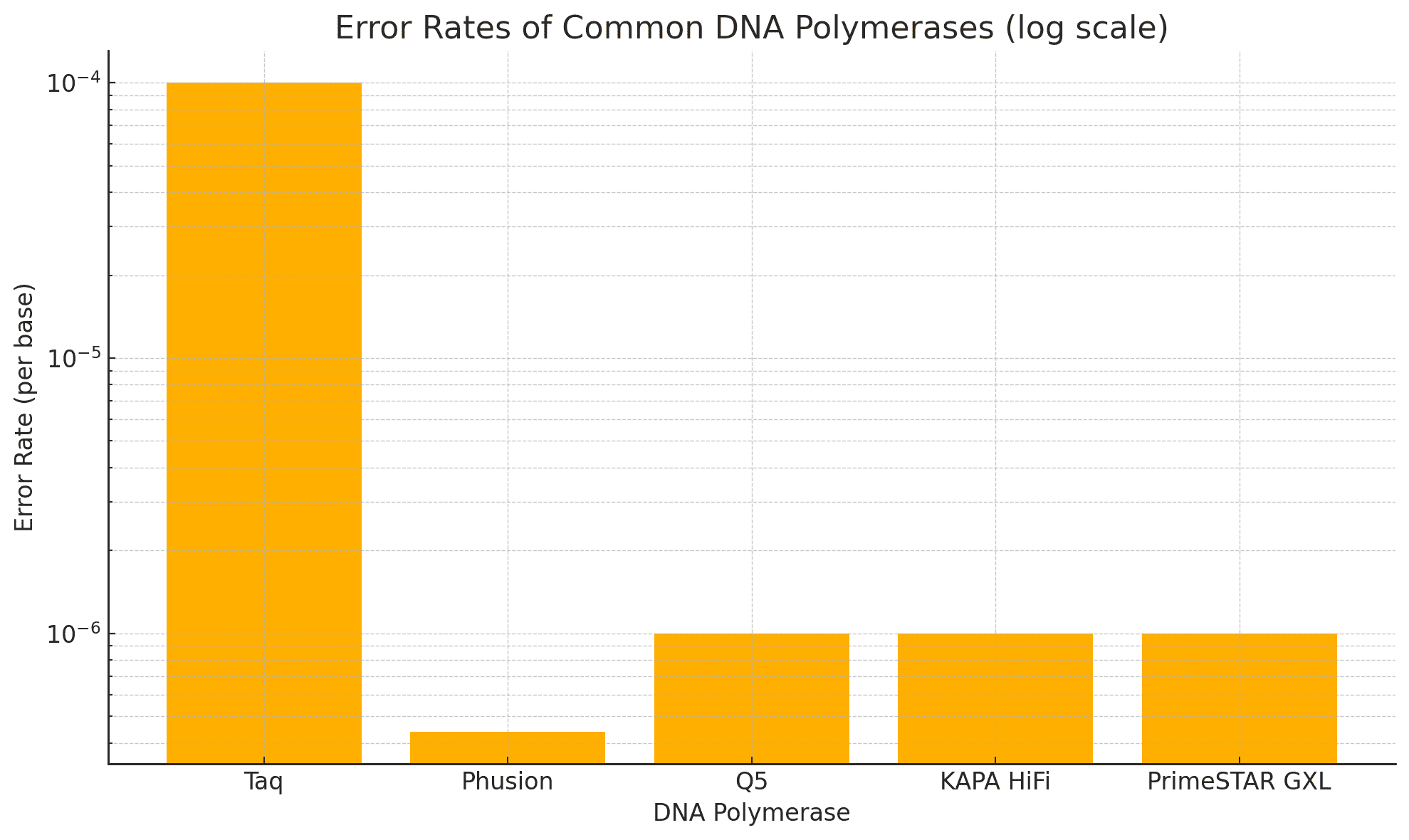
Error Rates: Standard vs. High-Fidelity Polymerases
| Enzyme Type | Error Rate (per base) | Proofreading Activity | Common Use |
|---|---|---|---|
| Standard Taq Polymerase | ~1 in 10⁴ | No | Routine PCR |
| High-Fidelity Enzymes | ~1 in 10⁶ to 10⁷ | Yes (3′→5′ exonuclease) | Library prep |
As described in NIH resources, high-fidelity polymerases like Q5®, Phusion®, and KAPA HiFi dramatically reduce base misincorporation and provide more even coverage across GC-rich or repetitive regions.
Further comparison studies by UC Berkeley and NC State confirm that when applied to complex genomic libraries, high-fidelity enzymes lead to greater mapping accuracy, lower duplication rates, and clearer read alignments.
Molecular Design of High-Fidelity Enzymes
High-fidelity enzymes incorporate a 3′ to 5′ exonuclease domain that actively scans the growing DNA strand for errors. When a misincorporated nucleotide is detected, the polymerase stalls, excises the error, and resumes synthesis with the correct base. This real-time proofreading minimizes accumulation of artifacts.
Moreover, many commercial enzymes include hot-start modifications, ensuring minimal non-specific activity during setup and initial temperature ramping. For example:
-
Q5 Hot Start High-Fidelity DNA Polymerase by NEB is ideal for long-read amplification and adapter ligation steps.
-
PrimeSTAR GXL by Takara Bio is optimized for difficult templates, such as high-GC genomic loci.

Critical PCR Steps in NGS Library Prep
1. Pre-Amplification of Low-Input DNA
Many library protocols begin with very small amounts of DNA or cDNA. High-fidelity polymerases ensure low amplification bias in such contexts University of Utah.
2. Adapter-Ligated Library Amplification
Incorrectly amplified adapter regions can lead to barcode cross-contamination, resulting in misassigned reads. Using proofreading enzymes helps retain correct adapter fidelity UCLA.
3. PCR-Based Target Enrichment
For amplicon-based NGS methods like 16S rRNA, amplicon panels, and exome capture, low error rates are essential to avoid false variant calls University of Minnesota.
Minimizing Error Accumulation: Tips and Best Practices
1. Use the Fewest Possible PCR Cycles
Overcycling increases chimeric and erroneous products. Guidelines from MIT suggest optimizing input DNA to keep cycle numbers <15 whenever possible.
2. Use UMI-Barcoded Primers
Unique Molecular Identifiers (UMIs) allow computational tools to deduplicate reads and remove PCR-induced variants Broad Institute.
3. Enzyme Buffer Optimization
High-fidelity enzymes are often buffer-specific. For instance, Q5 requires a high-salt buffer system to maintain fidelity in GC-rich templates University of Oregon.
4. Avoid Freeze-Thaw of Enzymes
Multiple freeze-thaw cycles can damage the proofreading domain. Store enzyme stocks in single-use aliquots NIH Protocol Library.
Real-World Application: Metagenomics and Rare Variant Studies
In studies of rare microbial taxa or ultra-low-frequency somatic mutations, accuracy is paramount. According to DOE Joint Genome Institute, even a single PCR-induced error can mimic a mutation present at <1% frequency, which may be falsely interpreted as a true subpopulation.
Use of enzymes like:
-
KAPA HiFi (low bias in high-complexity libraries)
-
AccuPrime Taq HiFi (optimized for multiplexed PCR)
has helped achieve high sensitivity in microbial ecology and environmental NGS applications.
Benchmarking Enzymes: How to Choose
Here is a chart from University of Wisconsin-Madison Biotech Center comparing popular high-fidelity enzymes:
| Enzyme | Error Rate | Max Amplicon Length | GC-Rich Tolerance | Hot Start |
|---|---|---|---|---|
| Q5 | ~10⁻⁶ | 20 kb | High | Yes |
| KAPA HiFi | ~10⁻⁶ | 15 kb | Moderate | Yes |
| PrimeSTAR GXL | ~10⁻⁶ | 30 kb | Excellent | Yes |
| Phusion | ~4.4×10⁻⁷ | 10 kb | Moderate | No |
Computational Error Suppression: The Bioinformatics Role
Once sequencing is complete, high-fidelity library preparation simplifies downstream analysis. Error suppression software like:
-
LoFreq NCBI
-
GATK HaplotypeCaller Broad Institute
-
UMI-tools Harvard
becomes more effective when input reads are clean, non-duplicated, and true to the template. High-fidelity enzymes increase base quality scores, reduce trimming artifacts, and improve alignment.
Conclusion
As NGS continues to evolve toward ultra-high resolution applications, the margin for technical errors becomes slimmer. By incorporating high-fidelity enzymes into the PCR stages of NGS library prep, researchers can significantly reduce the propagation of sequencing artifacts, improve confidence in variant analysis, and maintain the biological accuracy of their results.
Whether working on metagenomics, epigenetics, mutation profiling, or microbial evolution, enzyme selection is not just a protocol step—it is an essential decision point that influences every aspect of data quality.
For more information and detailed protocols, consult: